QC Visualization
1. Read-Level QC (Visualization)
The qc_visualization() function performs read-level
quality control visualization based on precomputed read count tables. It
serves as the R-side visualization companion to the bash-based QC step
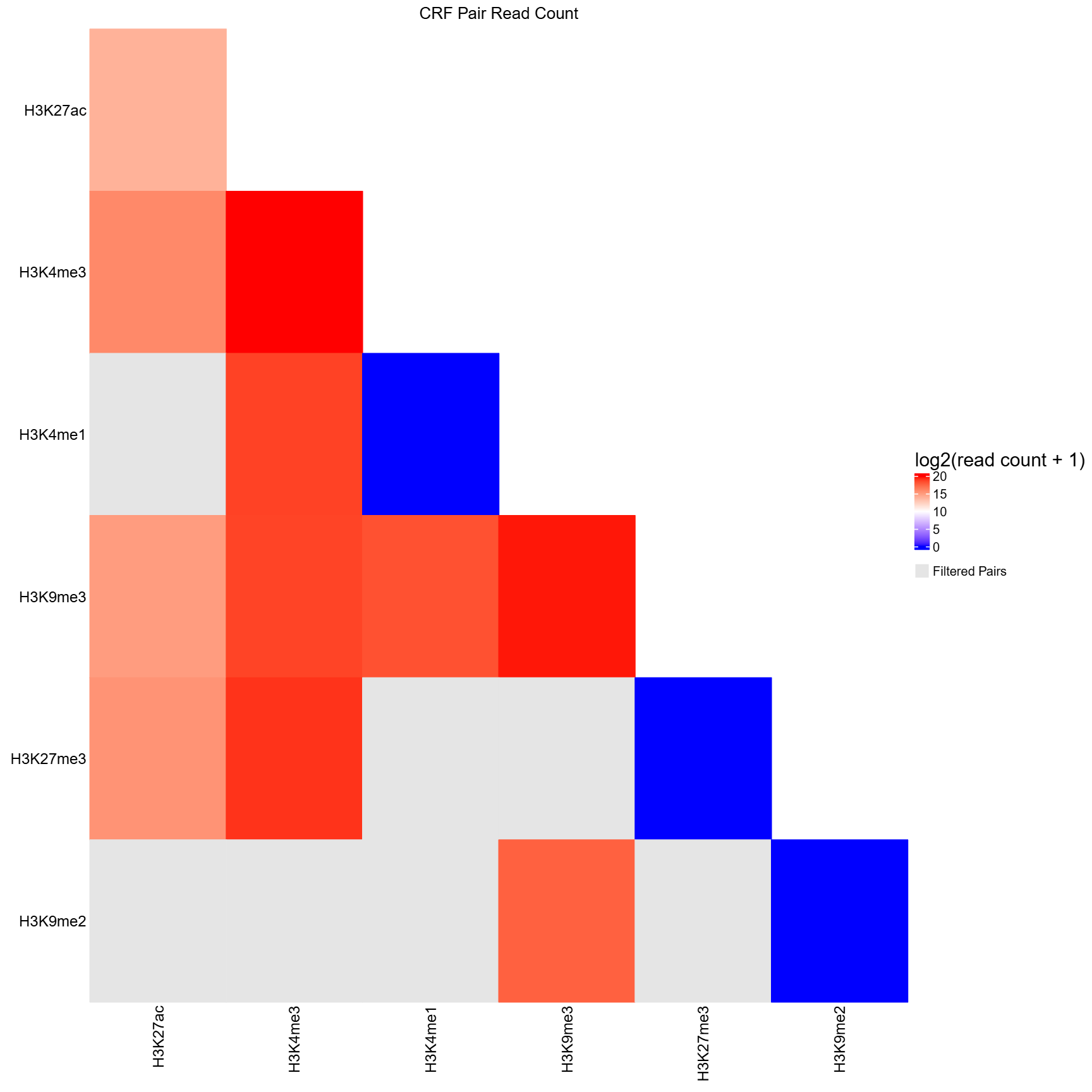
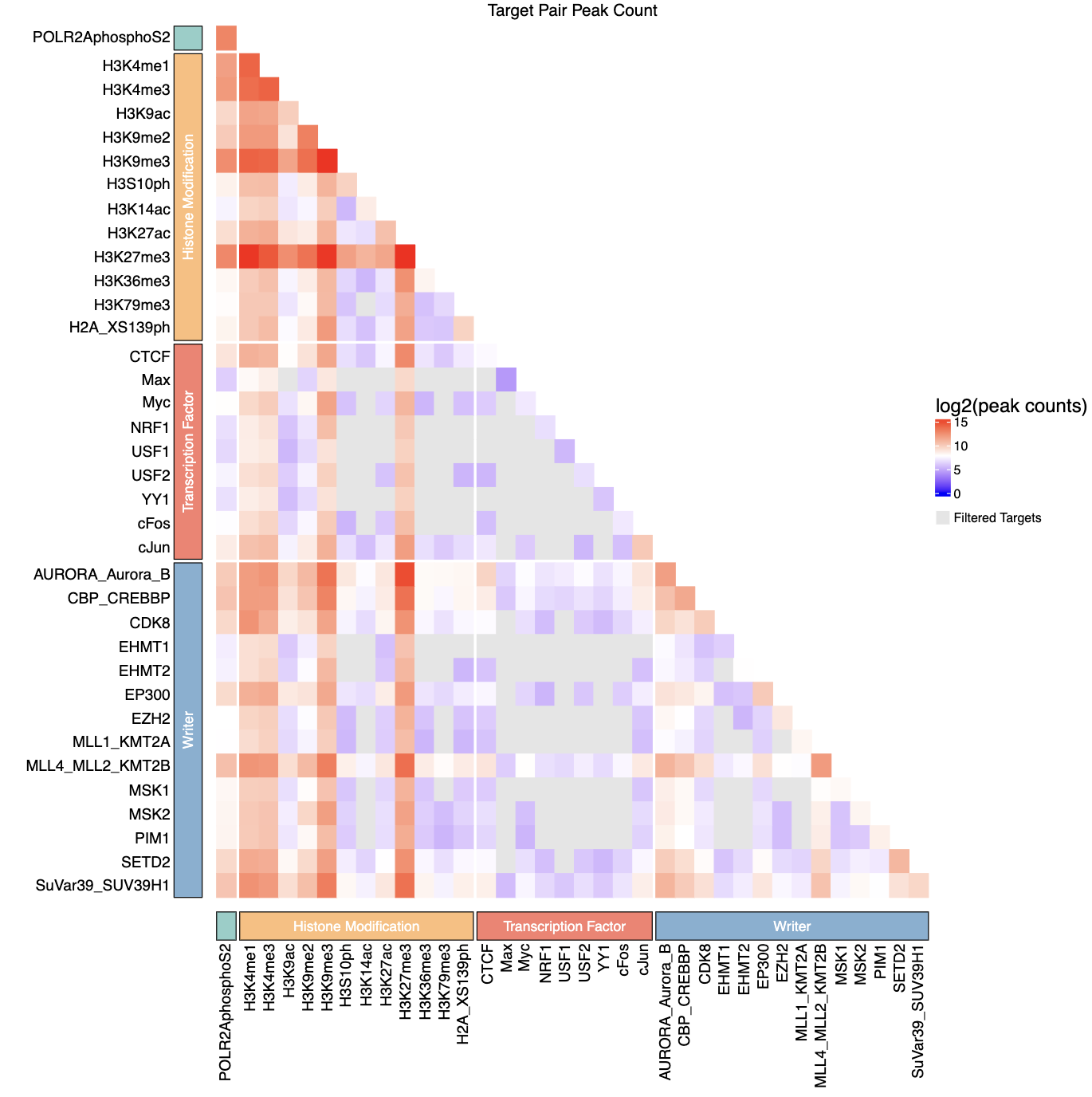
(qc.sh), transforming pairwise read counts into a
CRF-by-CRF heatmap for diagnostic inspection.
What this function does:
Unlike earlier integrated QC command, qc_visualization()
does not compute read counts or apply filtering itself.
Instead, it consumes TSV files generated upstream.
- Reads
all_qc.tsvcontaining CRF pair names (pair) and corresponding read counts (read_count). - Converts the pairwise table into a symmetric CRF-by-CRF matrix, filling only the lower triangle.
- CRF pairs absent from
all_qc.tsv(i.e., never sequenced) are marked asNAand displayed in grey. - Optionally reads
filtered_qc.tsvto further mark pairs that fail QC asNA(also grey). If not provided, all sequenced pairs are shown without QC filtering. - Applies a log2(read count + 1) transformation to stabilize dynamic range.
- Generates a publication-quality PDF heatmap summarizing sequencing depth across all CRF pairs.
- Optionally annotates CRFs by biological categories (e.g., histone marks, TFs) using a user-provided grouping CSV.
Parameters
| Parameter | Type | Default | Description | Example |
|---|---|---|---|---|
all_read_count_path
|
character | — |
Path to all_qc.tsv containing all CRF pairs and their read
counts
|
“qc/all_qc.tsv”
|
filtered_read_count_path
|
character | NULL |
Path to filtered_qc.tsv containing CRF pairs that pass QC.
If NULL, no QC filtering is applied; pairs absent from
all_read_count_path are still shown in grey.
|
“qc/filtered_qc.tsv”
|
out_dir
|
character |
“.”
|
Output directory for the heatmap PDF |
out_dir = “./qc”
|
split_pair_by
|
character |
“-”
|
Delimiter used to split CRF pair names into individual CRFs |
split_pair_by = “_“
|
group_csv
|
character |
NULL
|
Optional CSV defining CRF groupings for heatmap annotation |
“crf_groups.csv”
|
crf_col
|
character |
“crf”
|
Column name in group_csv specifying CRF identifiers
|
crf_col = “CRF”
|
category_col
|
character |
“category”
|
Column name in group_csv specifying CRF categories
|
category_col = “group”
|
Output Files
The function generates the following output files in the specified
out_dir:
- Peak count heatmap -
qc_heatmap.pdf(ifplot= TRUE)- PDF visualization of log2-transformed read counts for CRF-CRF pairs
- Lower triangle matrix showing read counts between CRF pairs
- Filtered (non-significant) pairs displayed in grey
- If
group_csvprovided, includes categorical annotations with colored blocks
Example Usage
library(multiEpiCore)
# ===== General Usage =======
# Define QC directory
qc_dir <- "qc"
# Optional: Create grouping CSV
crfs <- c("H3K4me1", "H3K4me3", "H3K9ac", "H3K9me2", "H3K9me3", "H3K27ac", "H3K27me3", "H3K36me3")
categories <- c(rep("Active", 3), rep("Repressive", 3), rep("Other", 2))
group_df <- data.frame(crf = crfs, category = categories)
write.csv(group_df, file.path(qc_dir, "crf_groups.csv"), row.names = FALSE)
# Run QC with grouping
qc_visualization(
all_read_count_path = file.path(qc_dir, "all_qc.tsv"),
filtered_read_count_path = file.path(qc_dir, "filtered_qc.tsv"),
out_dir = qc_dir,
split_pair_by = "-",
group_csv = file.path(qc_dir, "crf_groups.csv")
)
# ===== Test Data =======
qc_dirs <- list.dirs("qc", full.names = TRUE, recursive = FALSE)
for (qc_dir in qc_dirs) {
qc_visualization(
all_read_count_path = file.path(qc_dir, "all_qc.tsv"),
filtered_read_count_path = file.path(qc_dir, "filtered_qc.tsv"),
out_dir = qc_dir
)
}2. (Optional) Fragment Length Analysis
The frag_decomposition() function analyzes fragment
length distributions from BAM or BED files to characterize chromatin
structure patterns.
What this function does:
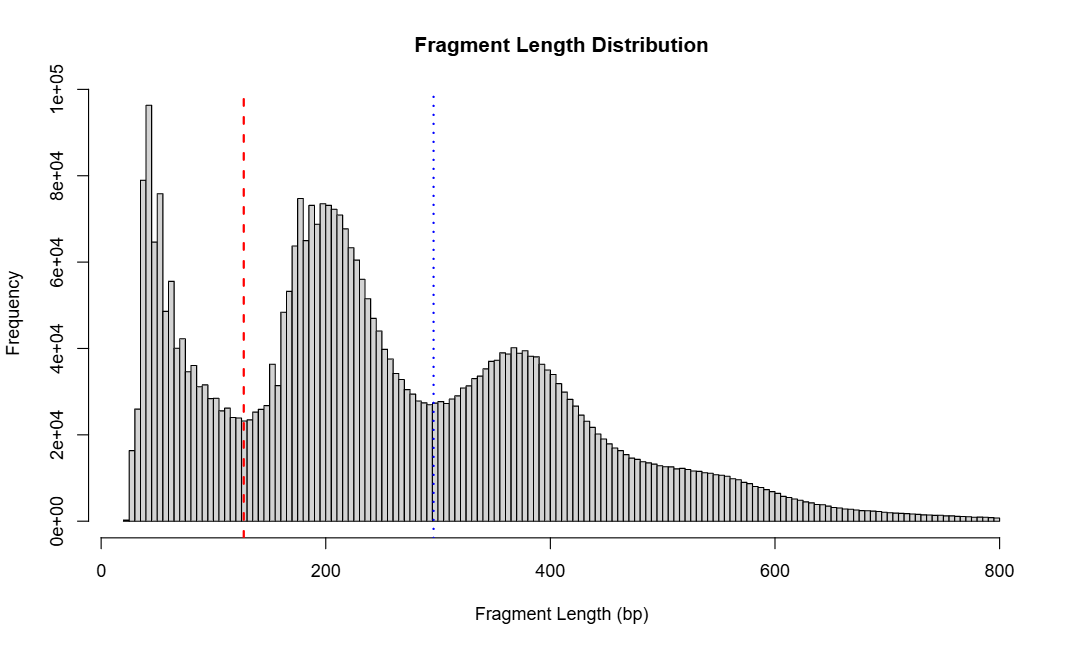
This function reads sequencing data files and extracts fragment lengths for all samples. It generates a combined histogram showing the overall fragment length distribution across all samples.
When detect_valley = TRUE, the function uses kernel
density estimation to identify two valley positions in the distribution:
the first valley (~150 bp) separates nucleosome-free regions from
mononucleosome-bound fragments, and the second valley (~300 bp)
separates mononucleosomes from dinucleosomes. These valleys serve as
thresholds to classify each fragment into one of three categories:
subnucleo (short fragments from open chromatin), monomer (single
nucleosome), or dimer+ (multiple nucleosomes).
The function then calculates the count and percentage of fragments in each category for every sample, providing a quantitative assessment of chromatin accessibility. All results are saved as a histogram PDF and a TSV file containing per-sample decomposition statistics.
Parameters
| Parameter | Type | Default | Description | Example |
|---|---|---|---|---|
file_path
|
character vector | — | Vector of BAM or BED file paths to analyze for fragment length distribution |
file_path = c(“sample1.bam”, “sample2.bam”)
|
out_dir
|
character |
“.”
|
Output directory |
out_dir = “./frag_analysis”
|
detect_valley
|
logical |
FALSE
|
If TRUE, detects valley positions in the fragment length distribution to classify fragments into subnucleo, monomer, and dimer+ categories |
detect_valley = TRUE
|
dens_reso
|
numeric |
2^15
|
Resolution for kernel density estimation; higher values produce smoother curves |
dens_reso = 2^16
|
dens_kernel
|
character |
“gaussian”
|
Kernel type for density estimation (options: “gaussian”, “epanechnikov”, “rectangular”, “triangular”, “biweight”, “cosine”) |
dens_kernel = “epanechnikov”
|
valley1_range
|
numeric vector |
c(73, 221)
|
Range to search for the first valley (NFR/mononucleosome boundary) |
valley1_range = c(100, 180)
|
valley2_range
|
numeric vector |
c(221, 368)
|
Range to search for the second valley (mono/dinucleosome boundary) |
valley2_range = c(250, 350)
|
Output Files
The function generates the following output files in the specified
out_dir:
- Fragment length histogram -
fragment_distribution.pdf- Shows the combined fragment length distribution across all samples
- If
detect_valley = TRUE, includes red and blue vertical lines marking the two detected valley positions - Visualizes the overall chromatin structure pattern
- Fragment decomposition data -
fragment_decomposition.tsv(only ifdetect_valley = TRUE)- Per-sample statistics table with the following columns:
pair: Sample name (filename without extension)total_count: Total number of fragmentssubnucleo_count/subnucleo_pct: Count and percentage of fragments < valley1 (nucleosome-free regions)monomer_count/monomer_pct: Count and percentage of fragments between valley1 and valley2 (mononucleosomes)dimer_plus_count/dimer_plus_pct: Count and percentage of fragments ≥ valley2 (dinucleosomes and higher)
- Per-sample statistics table with the following columns:
| pair | total_count | subnucleo_count | subnucleo_pct |
|---|---|---|---|
| H3K27ac-H3K27me3 | 42908 | 9422 | 21.9586091171809 |
| H3K27ac-H3K4me3 | 60028 | 11445 | 19.0661024855068 |
| H3K27me3-H3K4me1 | 1103829 | 266727 | 24.1637971098784 |
| H3K27me3-H3K4me3 | 569524 | 124800 | 21.9130361494862 |
| H3K27me3-H3K9me2 | 186552 | 39821 | 21.3457909858913 |
| H3K27me3-H3K9me3 | 881860 | 187616 | 21.275032318055 |
| H3K4me1-H3K4me1 | 283951 | 53164 | 18.7229486777648 |
| H3K4me1-H3K4me3 | 428026 | 84969 | 19.8513641694663 |
| H3K4me1-H3K9me3 | 301820 | 63064 | 20.8945729242595 |
| H3K4me3-H3K4me3 | 931528 | 187468 | 20.1247842254876 |
| H3K4me3-H3K9me2 | 79994 | 20079 | 25.1006325474411 |
| H3K4me3-H3K9me3 | 410770 | 96583 | 23.5126713245855 |
| H3K9me2-H3K9me2 | 111338 | 31467 | 28.2625877957211 |
| H3K9me2-H3K9me3 | 197180 | 47996 | 24.3412110761741 |
| H3K9me3-H3K9me3 | 796733 | 171545 | 21.531052435383 |
| pair | monomer_count | monomer_pct | dimer_plus_count | dimer_plus_pct |
|---|---|---|---|---|
| H3K27ac-H3K27me3 | 20203 | 47.084459774401 | 13283 | 30.956931108418 |
| H3K27ac-H3K4me3 | 21810 | 36.3330445791964 | 26773 | 44.6008529352969 |
| H3K27me3-H3K4me1 | 579365 | 52.4868435237704 | 257737 | 23.3493593663511 |
| H3K27me3-H3K4me3 | 274135 | 48.1340558080081 | 170589 | 29.9529080425057 |
| H3K27me3-H3K9me2 | 84384 | 45.2335005789271 | 62347 | 33.4207084351816 |
| H3K27me3-H3K9me3 | 428410 | 48.5802735128025 | 265834 | 30.1446941691425 |
| H3K4me1-H3K4me1 | 113860 | 40.0984676933696 | 116927 | 41.1785836288655 |
| H3K4me1-H3K4me3 | 176068 | 41.1348843294566 | 166989 | 39.013751501077 |
| H3K4me1-H3K9me3 | 132337 | 43.8463322510105 | 106419 | 35.25909482473 |
| H3K4me3-H3K4me3 | 341927 | 36.7060356747194 | 402133 | 43.169180099793 |
| H3K4me3-H3K9me2 | 34003 | 42.5069380203515 | 25912 | 32.3924294322074 |
| H3K4me3-H3K9me3 | 177366 | 43.1789079046668 | 136821 | 33.3084207707476 |
| H3K9me2-H3K9me2 | 40098 | 36.0146580682247 | 39773 | 35.7227541360542 |
| H3K9me2-H3K9me3 | 78349 | 39.734760117659 | 70835 | 35.924028806167 |
| H3K9me3-H3K9me3 | 332652 | 41.7520047493953 | 292536 | 36.7169428152217 |
Example Usage
library(multiEpiCore)
# ===== Test Data =======
bam_dirs <- list.dirs("bam", full.names = TRUE, recursive = FALSE)
for (bam_dir in bam_dirs) {
name <- basename(bam_dir)
bam_paths <- list.files(path = bam_dir, pattern = "\\.bam$", recursive = TRUE, full.names = TRUE)
frag_decomposition(
file_path = bam_paths,
out_dir = file.path("qc", name),
detect_valley = TRUE
)
}