Quality Control
1. Read-Level QC
The all_qc_with_plot() function performs quality control
on BAM/BED files and generates a log2-transformed peak count heatmap for
tag-tag pairs.
What this function does:
Performs quality control analysis on BAM or BED files
Calculates read counts and peak counts for all tag pairs
Filters samples based on read count percentiles
Generates a heatmap visualizing peak counts between tag pairs
Highlights filtered (non-significant) pairs in the heatmap
Supports optional tag grouping and categorical annotation
Saves QC summary files and visualization as outputs
Parameters
| Parameter | Type | Required | Description | Example |
|---|---|---|---|---|
file_path |
character vector | Yes | Vector of BAM or BED file paths for QC and visualization | file_path = c("sample1.bam", "sample2.bam") |
filtered_path |
character vector | No (default: NULL) | Optional vector of file paths for filtered QC matrix | filtered_path = c("filtered1.bam", "filtered2.bam") |
filtered_percentile |
numeric | No (default: 0.25) | Numeric value between 0 and 1 to filter the lowest-read-count samples | filtered_percentile = 0.25 |
output_path_dir |
character | No (default: NULL) | Directory to store output CSVs and PDF. If NULL, uses the directory of the first input file | output_path_dir = "./results" |
split_crf_by |
character | No (default: “-”) | Delimiter used to split tag pairs into individual TF names | split_crf_by = "-" |
save |
logical | No (default: TRUE) | If TRUE, writes CSV files and total reads summary | save = TRUE |
group_csv |
data frame | No (default: NULL) | Optional data frame defining tag groupings and categories for annotation | group_csv = data.frame(tag = c("YY1", "cJun"), category = c("Group1", "Group2")) |
tag_names |
character | No (default: “tag”) | Column name in group_csv for tag identifiers |
tag_names = "TF_name" |
category_names |
character | No (default: “category”) | Column name in group_csv for group categories |
category_names = "TF_family" |
target_pair_mapping_df |
data frame | No (default: NULL) | Optional data frame for renaming tag names in the heatmap matrix | target_pair_mapping_df = NULL |
Output Files
The function generates the following output files in the specified
output_path_dir:
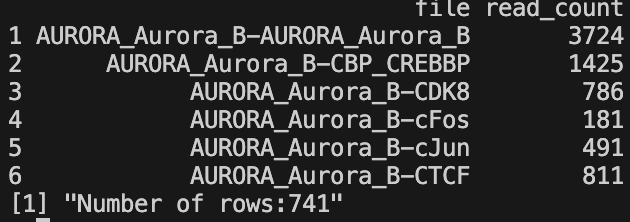
- All read counts CSV -
all_read_counts_bam.csvorall_read_counts_bed.csv- Contains read counts for all input files
- Two columns: file name and read count
- Generated only if
save = TRUE - File extension depends on input file type (BAM or BED)
- Filtered read counts CSV -
filtered_read_counts_bam.csvorfiltered_read_counts_bed.csv- Contains read counts for files passing the percentile filter
- Excludes samples in the lowest percentile based on
filtered_percentileparameter - Two columns: file name and read count
- Generated only if
save = TRUE 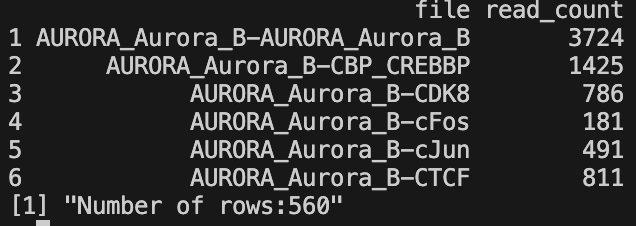
- Total reads summary -
total_reads.txt- Text file containing total read count across all samples
- Single numeric value
- Generated only if
save = TRUE 
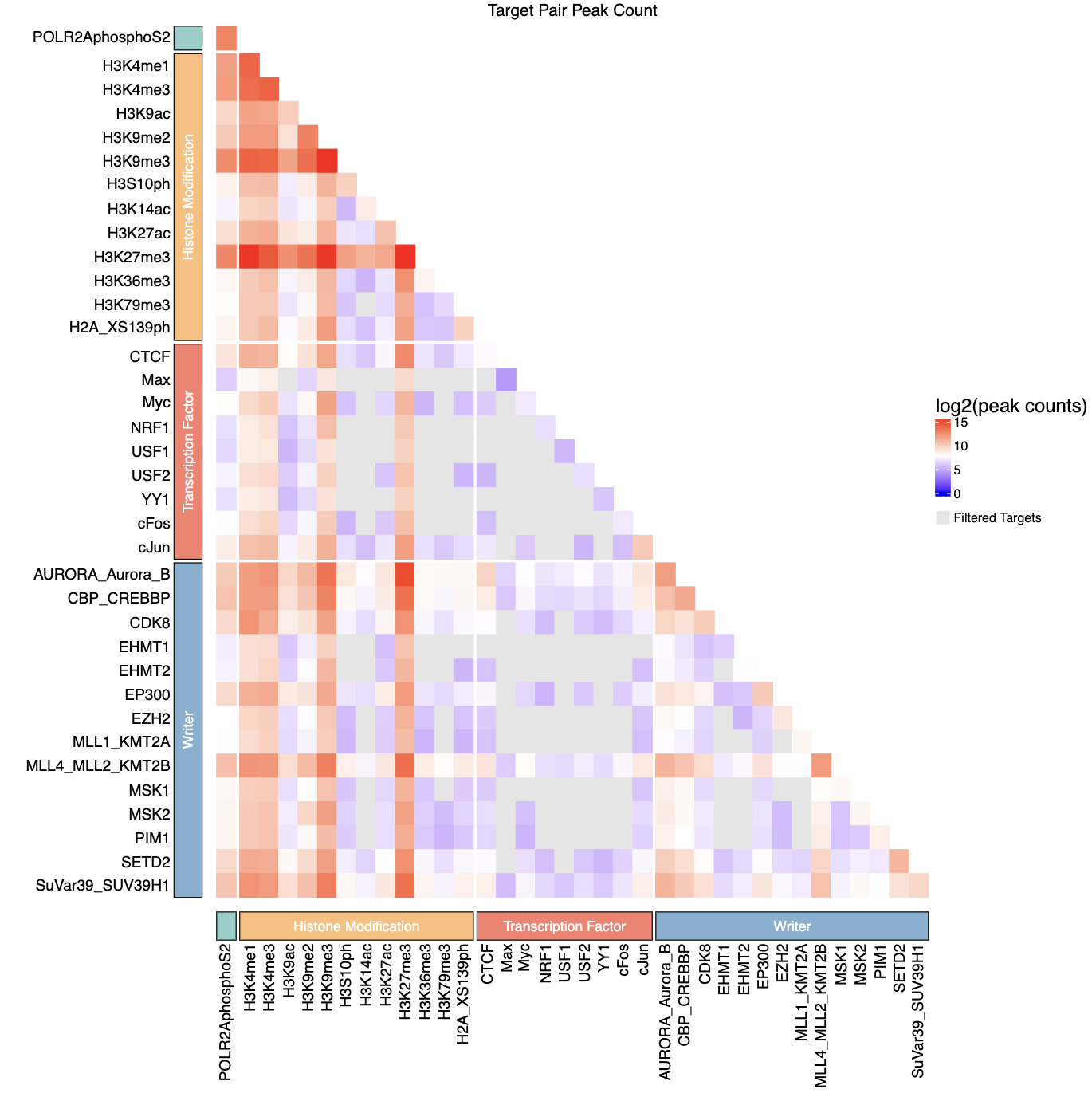
- Peak count heatmap -
AAA_test_qc_heatmap.pdf- PDF visualization of log2-transformed peak counts for tag-tag pairs
- Lower triangle matrix showing peak counts between transcription factor pairs
- Filtered (non-significant) pairs displayed in grey
- Color scale: blue (low) → white (medium) → red (high)
- If
group_csvprovided, includes categorical annotations with colored blocks - Dimensions: 12 × 12 inches
Example Usage
# bed file:
input_path <- "./dir_for_bed_files"
bed_files <- list.files(path = input_path, pattern = "\\.bed$", recursive = TRUE, full.names = TRUE)
# test csv
tags <- c("H3K4me1", "H3K4me3", "H3K9ac", "H3K9me2", "H3K9me3", "H3S10ph", "H3K14ac", "H3K27ac", "H3K27me3", "H3K36me3", "H3K79me3", "H2A_XS139ph", "POLR2AphosphoS2", "AURORA_Aurora_B", "CBP_CREBBP", "CDK8", "EHMT1", "EHMT2", "EP300", "EZH2", "MLL1_KMT2A", "MLL4_MLL2_KMT2B", "MSK1", "MSK2", "PIM1", "SETD2", "SuVar39_SUV39H1", "CTCF", "Max", "Myc", "NRF1", "USF1", "USF2", "YY1", "cFos", "cJun")
splits <- c(rep(1, 12), 2, rep(3, 14), rep(4, 9))
group_labels <- c("Histone Modification", "", "Writer", "Transcription Factor")
group <- group_labels[splits]
group_df <- data.frame(tag = tags, category = group)
library(epigenomeR)
all_qc_with_plot(file_path = bed_files, filtered_path = NULL, output_path_dir = "output/qc_heatmap", group_csv = group_df)Notes
The function automatically detects whether input files are BAM or BED format based on file extensions.
The
filtered_percentileparameter removes samples with the lowest read counts (e.g., 0.25 removes the bottom 25%).If
filtered_pathis provided, it overrides the percentile-based filtering for determining significant pairs in the heatmap.The heatmap displays only the lower triangle of the matrix, with filtered pairs shown in grey.
Peak counts are log2-transformed (log2(count + 1)) for visualization.
When
group_csvis provided, tags are sorted by category and displayed with colored block annotations.The function requires the following R packages:
ComplexHeatmap,latex2exp,circlize, andgrid.All file paths in
file_pathandfiltered_pathmust exist, or the function will fail.If
output_path_diris NULL, outputs are saved to the directory containing the first input file.
2. Fragment Length Analysis
The frag_decomposition() function performs a complete
fragment length analysis pipeline including quality control, valley
detection, and visualization for nucleosome positioning analysis from
BAM files.
What this function does:
Performs quality control filtering on BAM files based on coverage percentile
Computes fragment lengths from filtered paired-end BAM files
Detects two local minimum valleys for nucleosome fragment classification
Generates fragment length histograms with valley cutoff lines
Creates summary statistics report (valley positions, min/max fragment lengths)
Produces fragment decomposition data with percentages
Generates bar plots showing fragment distribution across samples
Parameters
| Parameter | Type | Required | Description | Example |
|---|---|---|---|---|
file_path |
character vector | Yes | Paths to BAM files to analyze | file_path = c("sample1.bam", "sample2.bam") |
save_dir |
character | Yes | Directory path to save all output files and plots | save_dir = "./fragment_analysis" |
frag_decomp_file |
character | No | Path to generated fragment decomposition file (returned by function) | Auto-generated |
filtered_percentile |
numeric | No | Percentile threshold for quality control filtering (default: 0.25, removes bottom 25%) | filtered_percentile = 0.25 |
dens_reso |
numeric | No | Resolution for kernel density estimation in valley detection (default: 2^15 = 32768) | dens_reso = 2^15 |
target_pair_mapping_df |
data frame | No | Optional data frame for mapping sample names to display names (default: NULL) | target_pair_mapping_df = df |
density_kernel |
character | No | Kernel type for density estimation (default: “gaussian”) | density_kernel = "gaussian" |
valley1_range |
numeric vector | No | Search range for first valley in bp (default: c(73, 221)) | valley1_range = c(73, 221) |
valley2_range |
numeric vector | No | Search range for second valley in bp (default: c(222, 368)) | valley2_range = c(222, 368) |
Output Files
The function generates the following output files in the specified
save_dir:
- Fragment length data -
premerge_all-qc_frag_lens.RData- Cached fragment length data from all filtered samples
- Numeric vector of fragment lengths
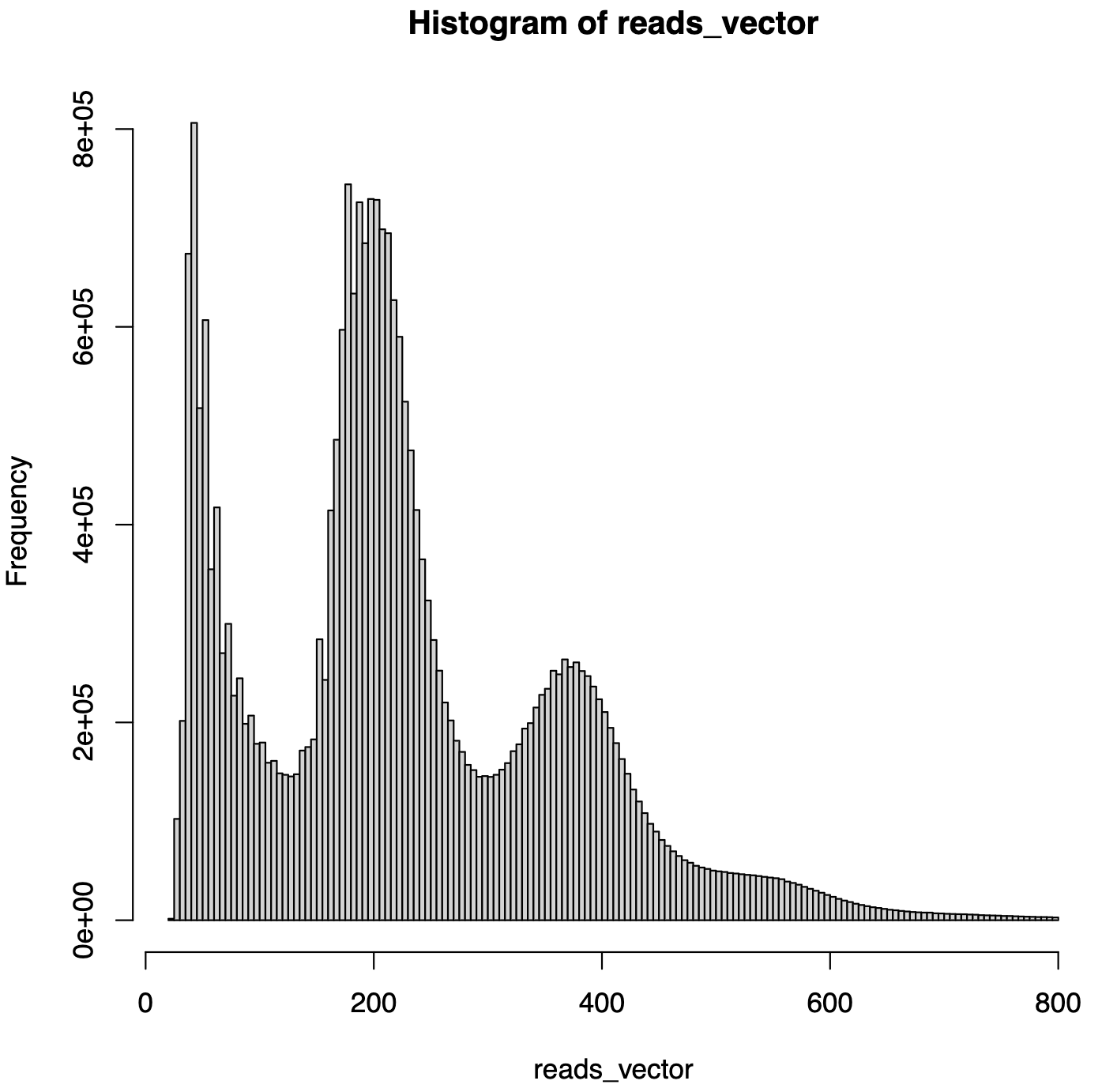
- Initial histogram -
premerge_frag_hist.pdf- Fragment length distribution histogram without valley cutoffs
- Shows overall fragment length distribution
- Summary report -
summary_report.csv- Contains key metrics:
local_min1: First valley position (bp)local_min2: Second valley position (bp)min_frag_len: Minimum fragment length (bp)max_frag_len: Maximum fragment length (bp)

- Contains key metrics:
- Histogram with cutoffs -
premerge_frag_hist_with_cutoff.pdf- Fragment length distribution with vertical lines at valley positions
- Used to visualize nucleosome fragment classification
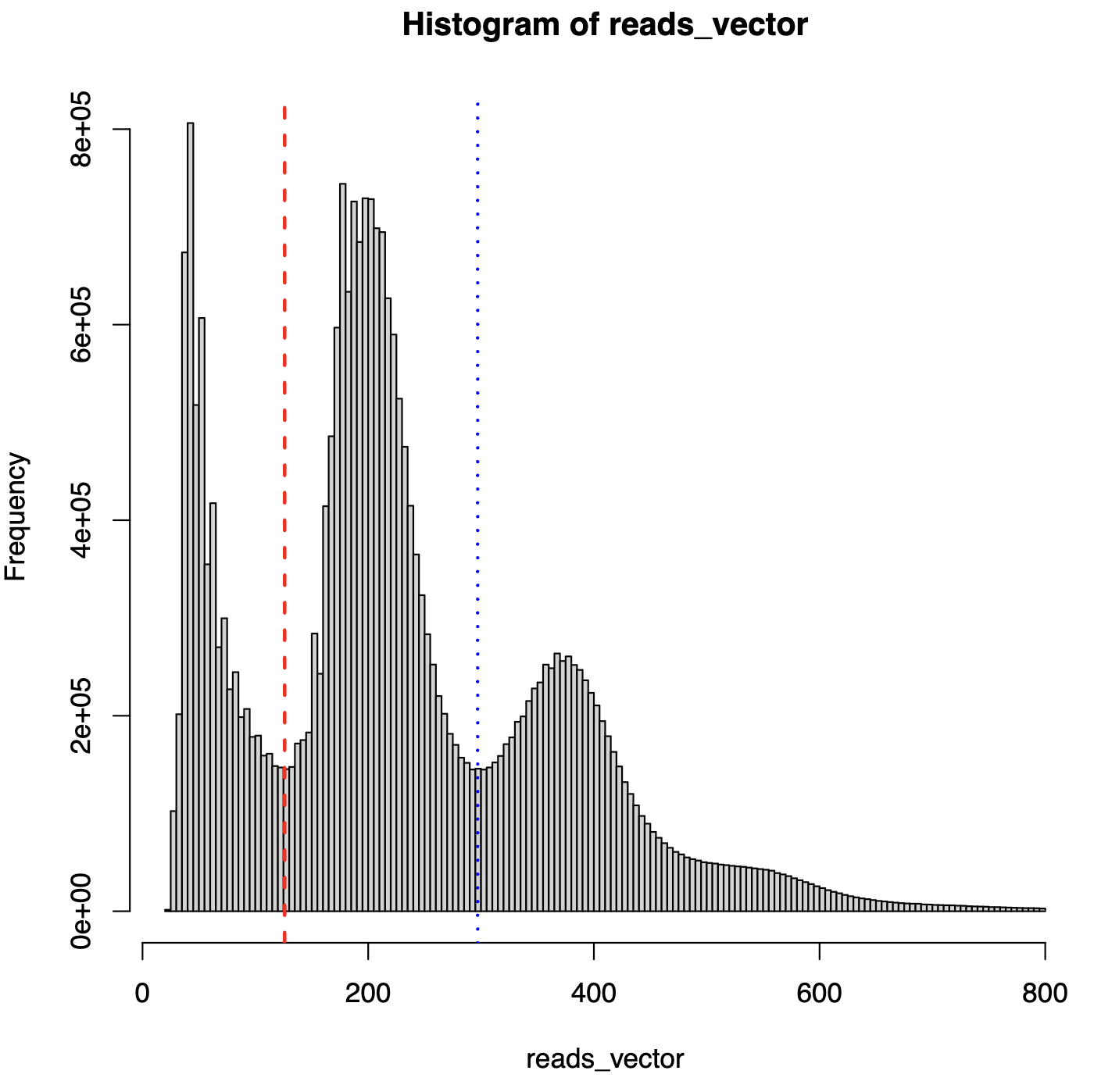
- Fragment decomposition data -
*_frag_decomp_with_perc.tsv- Per-sample fragment counts and percentages
- Classified by valley thresholds:
subnucleo,monomer,dimer+ 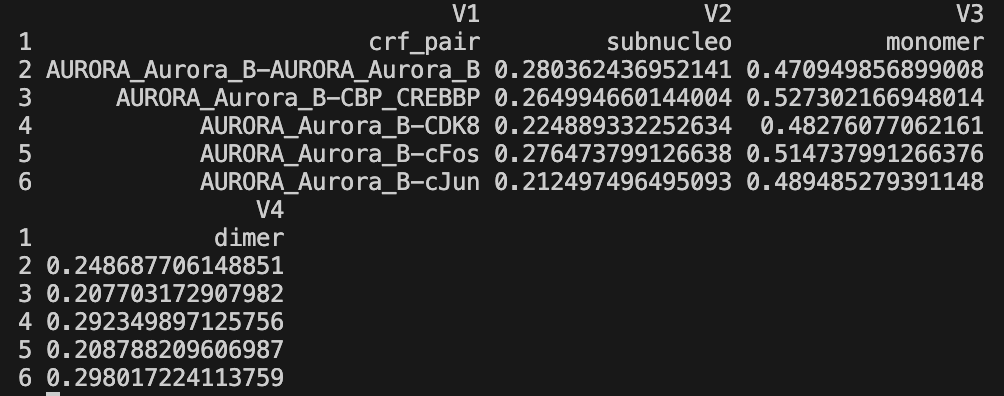
- Bar plots -
save_dir/barplots/*.pdf- Visual representation of fragment distribution across samples
- Separate plots for different fragment categories
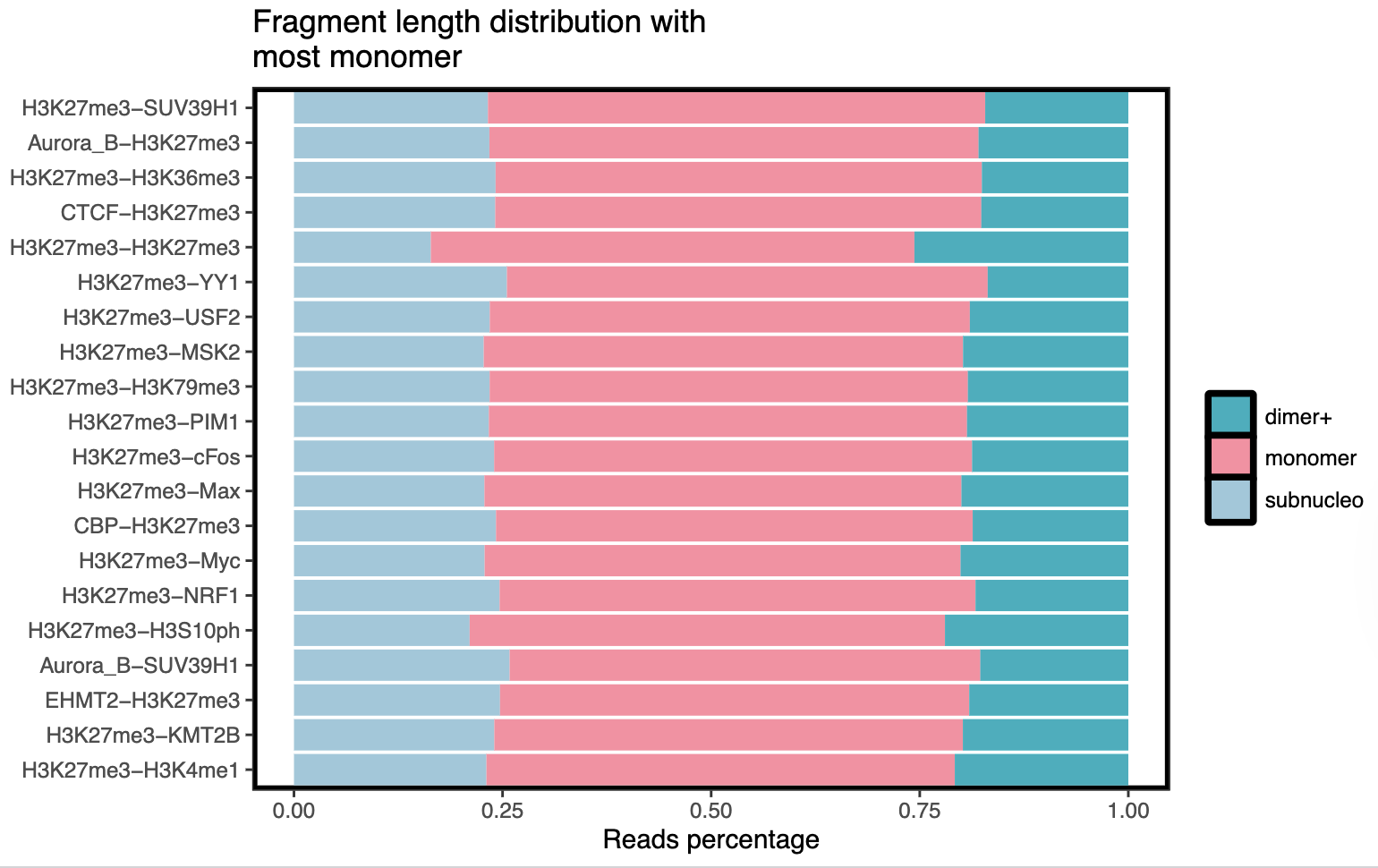
Example Usage
library(epigenomeR)
frag_decomposition(file_path = bam_files_vector, save_dir = "output/frag_decomposition")3. Split bam files in bash
## Use in bash
## change with your length of scen_list
#SBATCH --array=0-1
module load samtools
# setup (change)
thres_name="valley-V-qc"
scen_list=( "T" "V" ) # input dir name
scen_simple_name_list=( "T" "V" ) # output name
target_list=( "IgG_control" "H3K36me3" "H3K4me1"... )
load_root_dir="./data_align" # directory path for scen_list
save_root_raw_dir="output/frag_decomposition/split_bam"
# main function
run_frag_decomposition() {
scen=${scen_list[${SLURM_ARRAY_TASK_ID}]}
scen_simple_name=${scen_simple_name_list[${SLURM_ARRAY_TASK_ID}]}
save_root_dir=${save_root_raw_dir}/${thres_name}
mkdir -p "${save_root_dir}"
load_dir=${load_root_dir}/${scen}
mixed_dir=${save_root_dir}/${scen_simple_name}_mixed
subneucleo_dir=${save_root_dir}/${scen_simple_name}_sub
mononeucleo_dir=${save_root_dir}/${scen_simple_name}_mono
dineucleo_dir=${save_root_dir}/${scen_simple_name}_di
mkdir -p "${mixed_dir}" "${subneucleo_dir}" "${mononeucleo_dir}" "${dineucleo_dir}"
cp -r ${load_dir}/* "${mixed_dir}/"
for target_1 in "${target_list[@]}"; do
for target_2 in "${target_list[@]}"; do
if [ "${target_1}" \< "${target_2}" ] || [ "${target_1}" = "${target_2}" ]; then
mixed_filename="${target_1}-${target_2}.bam"
mixed_dir_filename=${load_dir}/${mixed_filename}
if [[ ! -f "${mixed_dir_filename}" ]]; then
echo "Warning: ${mixed_dir_filename} not found, skip."
continue
fi
samtools view -h ${mixed_dir_filename} | awk 'substr($0,1,1)=="@" || ($9>=24 && $9<=126) || ($9<=-24 && $9>=-126)' | samtools view -b > ${subneucleo_dir}/${mixed_filename}
samtools view -h ${mixed_dir_filename} | awk 'substr($0,1,1)=="@" || ($9>=127 && $9<=297) || ($9<=-127 && $9>=-297)' | samtools view -b > ${mononeucleo_dir}/${mixed_filename}
samtools view -h ${mixed_dir_filename} | awk 'substr($0,1,1)=="@" || ($9>=298 && $9<=800) || ($9<=-298 && $9>=-800)' | samtools view -b > ${dineucleo_dir}/${mixed_filename}
fi
done
done
}
run_frag_decomposition